Indikation
Efgartigimod (Vyvgart®) ist ein humanes IgG1-Ak-Fragment, das über die Bindung an den neonatalen Fc-Rezeptor (FcRn) wirkt. Der FcRn steht damit nicht mehr für das physiologische intrazelluläre Recycling von IgG zur Verfügung. IgG-Moleküle einschließlich pathologischer Antikörper wie die Acetylcholin-Rezeptor (AChR)-spezifischen Antikörper werden verstärkt lysosomal abgebaut und deren Serumkonzentration damit deutlich reduziert. In der für die Zulassung relevanten multizentrischen, randomisierten, doppelblinden und Placebo-kontrollierten Phase-III-Studie ADAPT wurde der primäre Studienendpunkt für die AChR-Antikörper-positive generalisierte Myasthenia gravis (AChR-Ak-positive gMG) erreicht. Dieser war definiert als klinisch signifikante Verbesserung des MG-ADL-Gesamtscores um ≥ 2 Punkte in mindestens 4 aufeinanderfolgenden Wochen im Vergleich zum Wert zu Anfang des Behandlungszyklus, wobei die erste Verbesserung nicht später als eine Woche nach der letzten Infusion des Zyklus festzustellen war. Efgartigimod (Vyvgart®) ist seit August 2022 als Zusatztherapie zur Standardbehandlung der AChR-Ak-positiven gMG zugelassen. Die subkutane Formulierung wurde in einer zehnwöchigen, multizentrischen, randomisierten, offenen Parallelgruppenstudie im Vergleich mit der o. g. intravenösen Therapie getestet, wobei eine sogenannte „Nicht-Unterlegenheit“ der subkutanen gegenüber der intravenösen Verabreichungsform nachgewiesen werden konnte (ADAPTsc-Studie). Die meisten Patienten (> 80 %) mit MG-ADL-Ansprechen zeigten bereits innerhalb der ersten 2 Wochen der Behandlung ein Ansprechen auf die Therapie mit Vyvgart subcutan®. Die subkutane Formulierung ist seit Mitte Dezember 2023 in Deutschland verfügbar. Jenseits der Standardtherapien liegen keine direkten prospektiven Vergleichsdaten zur Wirksamkeit für weitere in den Qualitätshandbüchern beschriebene MG-Therapieoptionen (z. B. Rituximab, Eculizumab, Ravulizumab) vor.
Die Entscheidung zur Therapie mit Efgartigimod sollte nach sorgfältiger Nutzen-Risiko-Abwägung unter Einbeziehung von Krankheitsaktivität, Therapierisiken und möglichen Therapiealternativen individuell auf Basis der Empfehlungen der DGN-Leitlinie zur Diagnostik und Therapie myasthener Syndrome getroffen werden. Den aktuellen Studiendaten folgend und entsprechend der aktuellen Zulassung und DGN-Leitlinie besteht die Indikation für die Add-on-Therapie mit Efgartigimod bei AChR-Ak-positiven gMG-Patienten mit (hoch-)aktivem Krankheitsverlauf unter Standardtherapie (Pyridostigmin, Immunsuppressiva inkl. Thymektomie), d. h.,
- die weiterhin alltagsrelevante myasthene Symptome haben
- die kein ausreichendes Ansprechen auf eine Immunsuppression zeigen, aber hinreichend hoch dosiert und hinreichend lange eingesetzt wurden
- die aufgrund von Medikamentennebenwirkungen die bisherige Therapie abbrechen
- die wiederholt mit intravenösen Immunglobulinen oder Plasmaaustauschverfahren behandelt werden mussten und/oder
- intensivmedizinische Therapien wegen schwerer myasthener Exazerbationen/Krisen erlitten haben.
Es wird empfohlen, die Therapieentscheidung in Absprache mit einem in dieser Therapie und mit der Myasthenie erfahrenen Zentrum (z. B. einem integrierten Myasthenie Zentrum) zu treffen und die Patienten in einem spezifischen Krankheitsregister (z. B. dem Deutschen Myasthenie Register) zu erfassen. Bei der Wahl zwischen intravenöser und subkutaner Anwendung spielt vor allem der Patientenkomfort eine wichtige Rolle. Die subkutane Anwendung in der Häuslichkeit ist nach einer Schulungsphase durch den Patienten selbst möglich und bietet damit eine höhere Patientenautonomie.
Kontraindikationen
Eine absolute Kontraindikation besteht bei…
- Hypersensitivität gegenüber dem Wirkstoff oder einem der sonstigen Bestandteile des Arzneimittels.
- Hypogammaglobulinämie bzw. IgG-Mangel < 2 g/l.
Eine relative Kontraindikation besteht bei…
- Schwangerschaft und Stillzeit.
- akuten und chronischen systemischen Infektionen.
Dosierung
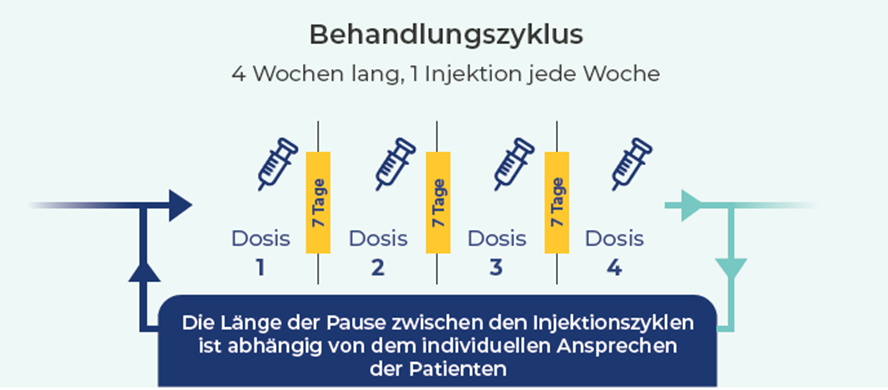
Efgartigimod wird als intravenöse Infusion gewichtsadaptiert mit 10 mg/kg Körpergewicht (KG) über 60 Minuten unter Verwendung eines Inline-Filters verabreicht. Ab einem KG von 120 kg dürfen max. 1200 mg infundiert werden. Aktuell erfolgt die Therapie entsprechend der ADAPT-Studie (inkl. der Open-Label-Extension-Phase ADAPT+) in Zyklen. Ein Zyklus besteht aus 4 Infusionen, die im wöchentlichen Abstand verabreicht werden (Tag 0, 7, 14, 21). Danach folgt ein infusionsfreies Intervall von mindestens 4 Wochen. In Abhängigkeit des klinischen Verlaufs kann anschließend ein erneuter Zyklus erfolgen, wobei die Intervalle flexibel sein können.
Die subkutane Therapie unterscheidet sich von der intravenösen Therapie nur in der subkutanen Applikation und der gewichtsunabhängig einzusetzenden Dosis. Seit März 2025 steht Vyvgart® als gebrauchsfertige Fertigspritze zur Verfügung und ersetzt die bisherige Durchstechflasche für die subkutane Anwendung. Die Fertigspritze enthält 1000 mg Efgartigimod alfa in 5 ml (200 mg/ml) und wird als fixe, gewichtsunabhängige Dosis verabreicht. (Die bisher beschriebene Zubereitung aus der Durchstechflasche (1000 mg in 5,6 ml, 180 mg/ml) mit Transferkanüle und geflügeltem Infusionsset entfällt für die subkutane Anwendung.) Dabei erfolgt die Therapie entsprechend der ADAPTsc-Studie (inkl. der Open-Label-Extension-Phase ADAPTsc+) ebenfalls in den aus der Infusionstherapie bekannten Regeln für die Zyklen. Für die ersten 5 Gaben von Vyvgart® Injektionslösung muss eine Interventionsmöglichkeit bei Injektions- oder Überempfindlichkeitsreaktionen sichergestellt sein, sodass diese Gaben im Krankenhaus bzw. in einer Praxis / einem Ambulanzzentrum stattfinden müssen. Ab der 6. Injektion können Patienten oder Pflegepersonen das Arzneimittel nach ausreichender Schulung zur subkutanen Injektionstechnik eigenständig zu Hause verabreichen.
Die Injektion des gesamten Arzneimittels dauert ca. 20–30 Sekunden.
Zur Beurteilung des klinischen Verlaufs sollte in erster Linie der MG-ADL-Score eingesetzt werden. Im Falle einer Verschlechterung sollte zeitnah ein neuer Zyklus unter Berücksichtigung des vierwöchentlichen infusionsfreien/injektionsfreien Intervalls erfolgen. Die Verschlechterung sollte messbar sein, aber nicht den Ausgangszustand vor dem jeweiligen Zyklus erreichen. Falls eine Gabe nicht stattfinden kann/konnte, sollte diese möglichst innerhalb von ± 3 Tagen nachgeholt werden. Wenn eine Dosis um mehr als 3 Tage verschoben werden muss, sollte die Dosis nicht gegeben werden, um sicherzustellen, dass die Gabe von zwei aufeinanderfolgenden Dosen im Abstand von mindestens 3 Tagen stattfindet. Bei klinischen Infektzeichen muss ggf. die Infusion ausgesetzt und die Wiederaufnahme der Infusion(en) nach Genesung von der Infektion muss individuell entschieden werden.

SUBKUTANE ANWENDUNG – FERTIGSPRITZE
Pharmakokinetik
- Efgartigimod wird durch proteolytische Enzyme in kleine Peptide und Aminosäuren abgebaut. Bei Patienten mit MG betrug die mittlere Clearance 0,108 L/h. Das molekulare Gewicht von Efgartigimod beträgt ca. 54 k. Dies bezieht sich auf Moleküle, die an der Grenze zur renalen Elimination liegen. Die mittlere Eliminationshalbwertszeit liegt bei 80–120 Stunden (3–5 Tage).
- Die Efgartigimod-Dosis war in der ADAPT-Zulassungsstudie limitiert auf 10 mg/kg KG bei Patienten bis 120 kg sowie bei Patienten mit > 120 kg KG auf eine maximale Dosis von 1200 mg/Infusion. In der ADAPT-Studie hatten 5 Patienten (3 %) ein Körpergewicht von > 120 kg, das mittlere Gewicht betrug 76,5 kg. Das KG hatte keinen Einfluss auf das Ausmaß der IgG-Reduktion.
- Die Pharmakokinetik wurde nicht vom Alter (19–78 Jahre), Geschlecht oder der ethnischen Abstammung beeinflusst.
- Es liegen begrenzte Daten zur Sicherheit und Wirksamkeit bei Patienten mit leichter Nierenfunktionsstörung vor; bei Patienten mit leichter Nierenfunktionsstörung ist keine Dosisanpassung erforderlich. Bei Patienten mit mäßiger Nierenfunktionsstörung sind die Daten zur Sicherheit und Wirksamkeit sehr begrenzt, und bei Patienten mit schwerer Nierenfunktionsstörung liegen keine Daten zur Sicherheit und Wirksamkeit vor.
- Es liegen keine pharmakokinetischen Daten bei Patienten mit Leberinsuffizienz vor. Leberfunktionsparameter blieben in den pharmakokinetischen Analysen unbeeinflusst.
- Efgartigimod kann als IgG-Antikörper potenziell in die Muttermilch übergehen und die Plazentaschranke passieren. Da davon auszugehen ist, dass Efgartigimod die mütterlichen Antikörperspiegel senkt und außerdem die Übertragung mütterlicher Antikörper auf den Fetus hemmt, ist eine Verringerung des passiven Schutzes des Neugeborenen zu erwarten.
Pharmakodynamik
- Efgartigimod senkt als FcRn-Antagonist die IgG-Gesamtkonzentration genauso wie die pathogenen Auto-Ak vom IgG-Typ, wobei kein Unterschied hinsichtlich der Subtypen IgG1 bis IgG4 besteht.
- In den ADAPT-Studien wurde die maximale Abnahme des Gesamt-IgG-Spiegels bei ca. 60 % der Patienten eine Woche nach der letzten Infusion bzw. Injektion des ersten Behandlungszyklus erreicht und normalisierte sich wieder auf das Ausgangsniveau ca. 9 Wochen nach der letzten Infusion bzw. Injektion. Die gleichen Effekte wurden für alle IgG-Subtypen beobachtet.
- Parallel hierzu zeigte sich eine Abnahme des AChR-Ak-Spiegels mit ebenfalls einer max. Abnahme von ca. 60 % der Patienten eine Woche nach der Infusion/Injektion, wobei ca. 7 Wochen nach der letzten Infusion bzw. Injektion wieder das Ausgangsniveau erreicht wurde. Diese Veränderungen zeigen sich hoch reproduzierbar in den nachfolgenden Zyklen.
- Efgartigimod hat keinen Einfluss auf die Serumkonzentrationen von IgA, IgM, IgD und IgE. Der Albumin-Spiegel kann invers zur Abnahme der IgG-Konzentration etwas ansteigen.
- Erste Daten zeigen keine offensichtlichen Auswirkungen von Antikörpern gegen Efgartigimod alfa auf die klinische Wirksamkeit oder Sicherheit oder auf die Pharmakokinetik und pharmakodynamischen Parameter.
Diagnostik || vor Therapiebeginn
Anamnese und klinische Untersuchung zu möglichen Kontraindikationen
Durch Anamnese und klinische Untersuchung sollten gezielt vor jeder Efgartigimod-Gabe mögliche Kontraindikationen (insbesondere Infektionen) ausgeschlossen werden. Die, wenn auch selten, im Kontext der MG zu beobachtenden Immundefizienz-Syndrome, insbesondere solche mit Hypogammaglobulinämie, sollten vor der erstmaligen Gabe von Efgartigimod ausgeschlossen werden. Anamnese und Untersuchung müssen detailliert dokumentiert werden (obligat). Bei Patienten mit aktiver systemischer Infektion sollte der Therapiebeginn verschoben werden, bis die Infektion remittiert ist (obligat). Aufgrund der Gefahr der infektassoziierten Verschlechterungen der MG sollte jedoch eine längere Therapiepause vermieden und ggf. die Efgartigimod-Therapie unter antiinfektiver Behandlung eingeleitet werden.
Labor-Basisprogramm
- Routinelaborparameter: Die Bestimmung des Differentialblutbilds, der Leberwerte (GOT, GPT, GGT, Bilirubin) und der Nierenwerte (Kreatinin) vor Therapiebeginn ist obligat.
- Die quantitative IgG-Bestimmung ist fakultativ, kann aber hilfreich sein, um eine Hypogammaglobulinämie zu vermeiden. Ein verbindlicher Schwellenwert existiert nicht. Orientiert man sich an der aktuellen Studienliteratur, sind Werte VOR Beginn der ersten Infusion eines jeden Zyklus von 4 g/l klinisch die untere Grenze.
- Entzündungs- und Infektionsparameter: Vor der Einstellung auf Efgartigimod sollte ein Screening auf akute Entzündungen (inkl. CRP und Urinstatus) erfolgen (obligat). Weiterführende Abklärungen auf chronische und virale Infektionen (Lues, HBV, HCV, HIV) können erfolgen (fakultativ). Bei V. a. Tbc in der Vorgeschichte oder Personen, die in Gebieten mit höherer Tbc-Prävalenz leben bzw. Kontakt zu Tbc-Erkrankten haben, sollte auf eine Tbc-spezifische Immunreaktion untersucht werden (mittels Tbc-spezifischem ELISPOT oder Interferon-Release-Test, z. B. Quantiferon®) (fakultativ) und bei positivem Testergebnis die Gefahr einer Tbc-Reaktivierung abgeklärt werden (Röntgen-Thorax und ggf. weitere Diagnostik) (fakultativ). Zudem sollte überprüft werden, ob Immunität gegen das Varizella-Zoster-Virus vorhanden ist (fakultativ).
- Schwangerschaftstest: Bei Patientinnen im gebärfähigen Alter muss ein negativer Schwangerschaftstest vorliegen oder die Schwangerschaft anderweitig sicher ausgeschlossen sein (obligat).
Dokumentierte Aufklärung der Patienten über Therapie und Risiken
Eine standardisierte Aufklärung über Risiken und Nutzen der Efgartigimod-Therapie und eine schriftliche Einwilligungserklärung des Patienten sind vor Behandlungsbeginn obligat.
Es sollte speziell auf ein erhöhtes Risiko von Infektionen sowie auf Vorsichtsmaßnahmen (sofortige ärztliche Vorstellung und ggf. früher Beginn einer entsprechenden Antibiotikatherapie) ausführlich eingegangen werden. In der ADAPT-Studie wurde eine erhöhte Rate an Atemwegsinfekten sowie Harnwegsinfekten beobachtet. Darüber hinaus sollte über das Auftreten von Infusionsreaktionen sowie eine erhöhte Rate von Kopfschmerzen aufgeklärt werden.
Bei subkutaner Anwendung sollte speziell auf ein erhöhtes Risiko von Injektionsreaktionen im Rahmen der ersten Anwendungen eingegangen werden (Hautausschlag, Pruritus). Die Patienten sollten während der ersten 5 Gaben und eine Stunde danach auf klinische Anzeichen und Symptome von Injektionsreaktionen und anaphylaktischen Reaktionen überwacht werden. Im Falle des Auftretens einer Reaktion müssen geeignete unterstützende Maßnahmen eingeleitet werden.
Vortherapien || Abstand und Maßnahmen
In den ADAPT-Studien wurde Efgartigimod als Add-on-Therapie eingesetzt. Die meisten Patienten wurden in ihrem vorangegangenen Krankheitsverlauf entsprechend den Leitlinienempfehlungen neben Pyridostigmin mit mindestens 2 der Standardimmuntherapeutika Kortikosteroiden, Azathioprin, Mycophenolatmofetil, Ciclosporin A, Tacrolimus, Methotrexat, Cyclophosphamid oder Rituximab behandelt. Ebenso war ein Teil der Patienten thymektomiert und wurde bei Exazerbationen bzw. myasthenen Krisen mittels intravenöser Immunglobuline und/oder apharetischer Verfahren behandelt. Im Regelfall sollte Efgartigimod als Zusatztherapie zur Standardtherapie eingesetzt werden. Eine Therapiepause der Standardtherapie ist weder notwendig noch sinnvoll. Ein Wechsel der immunsuppressiven Standardtherapie, z. B. wegen Nebenwirkungen, kann im Verlauf erfolgen und sollte von Laboruntersuchungen zur Erfassung von Komplikationen (z. B. Lympho-/Leukopenien, Leberwerterhöhungen) flankiert werden (fakultativ).
Während der Infusion/Injektion
Infusion
Die Verabreichung von Efgartigimod kann potenziell zu einer akuten Infusionsreaktion oder anaphylaktischen Reaktionen führen. Die Therapie soll daher nur unter entsprechenden Kautelen und engmaschiger Überwachung von erfahrenem medizinischen Fachpersonal durchgeführt werden (obligat). Die Infusionen müssen über 60 Minuten über eine Venenverweilkanüle mit sicherer intravenöser Gabe und über einen In-line-Filter durchgeführt werden (obligat). Die Patienten sollen während und eine Stunde lang nach der Infusion überwacht werden. Falls während der Verabreichung eine Nebenwirkung auftritt, kann die Infusion nach Ermessen des Arztes verlangsamt oder abgesetzt werden. Wenn die Infusion verlangsamt wird, sollte die Gesamtinfusionsdauer bei Erwachsenen zwei Stunden nicht überschreiten. Bei schweren Reaktionen muss die Infusion gestoppt und eine symptomatische Therapie eingeleitet werden.
Obwohl die vorliegenden Daten für ein sehr gutes Sicherheitsprofil sprechen, sollte der erste Zyklus in einem entsprechend aufgestellten Krankenhaus durchgeführt werden, wobei ein ambulantes Setting ausreichend ist. Begründend dafür ist, dass es sich um eine neue Therapie handelt. Die weiteren Behandlungszyklen sollten im ambulanten Setting bzw. niedergelassenen Bereich durch entsprechend erfahrene Kollegen unter den o. g. Kriterien durchgeführt werden.
Injektion
Für die subkutane Anwendung gelten ähnliche Empfehlungen. Dabei darf jeweils nur die Vyvgart®-Injektionslösung für die subkutane Gabe und die Vyvgart®-Infusionslösung für die intravenöse Gabe angewendet werden. Die Injektion erfolgt subkutan im Unterleibsbereich, mindestens 5 cm vom Nabel entfernt, bei jeder Gabe an einer neuen Stelle. Die Haut an der Injektionsstelle muss unauffällig sein (keine Injektion in Narben, Muttermale, gereizte, gerötete, infizierte oder verhärtete Haut). Die Nadel wird in einem Winkel von 45–90° eingeführt; der Kolben wird langsam bis zum Anschlag gedrückt. Bei aseptischer Arbeitsweise ist die Fertigspritze nach dem Öffnen sofort zu verwenden. Die Injektion soll langsam über einen Zeitraum von 20–30 Sekunden hinweg subkutan erfolgen. Die ersten 5 Injektionen sollten an einem erfahrenen Zentrum erfolgen, alle weiteren Gaben kann der Patient eigenständig in der Häuslichkeit durchführen. Die Patienten sollten während der Gabe und 30 Minuten danach auf klinische Anzeichen und Symptome von Injektionsreaktionen überwacht werden. Im Falle einer Injektionsreaktion sollten Folgegaben abhängig von der klinischen Bewertung mit den behandelnden Ärzten kritisch evaluiert werden.
Lagerung
- Im Kühlschrank aufbewahren (2–8 °C), in der Originalverpackung (Lichtschutz).
- Mindestens 30 Minuten vor der Injektion aus dem Kühlschrank nehmen, damit die Lösung Raumtemperatur annehmen kann.
- Nicht einfrieren.
- Ungeöffnete Fertigspritzen können bei Bedarf bei Raumtemperatur im Originalkarton gelagert werden (max. 30 °C, max. 1 Monat nach Entnahme aus dem Kühlschrank oder bis zum Verfallsdatum – je nachdem, was zuerst eintritt).
- Haltbarkeit: 24 Monate.
Mittel zur Behandlung möglicher anaphylaktischer und/oder schwerer Reaktionen müssen im Rahmen der Vyvgart-Gaben verfügbar und das Infusionsteam hinsichtlich der Behandlung von anaphylaktischen und/oder schweren Infusionsreaktionen geschult sein (obligat). Ein uneingeschränkter Zugang zu einer intensivmedizinischen Versorgungs- und Behandlungseinheit im eigenen Haus oder im nächstgelegenen Krankenhaus ist nach der Erstversorgung einer schweren Infusions- oder allergischen Reaktion erforderlich (obligat). Bezüglich der Handhabung und Herstellung der Infusionslösung muss die Fachinformation herangezogen werden.
Monitoring
Klinisch-neurologische Kontrolle
Vor jeder Medikamenten-Gabe sollen Patienten gezielt nach Zeichen einer Infektion befragt bzw. untersucht werden (obligat). Vor der ersten und vierten Infusion während eines Zyklus sollte mindestens der myastheniespezifische Score zu Aktivitäten des täglichen Lebens (MG-ADL-Score) erhoben werden, wobei begleitend die Bestimmung des quantitativen Myasthenia gravis (QMG)-Scores empfohlen wird. Weitere aussagekräftige Scores sind der MG-QoL15r und MGC (fakultativ). Damit lässt sich sowohl das Therapieansprechen als auch eine im Verlauf zu erwartende Verschlechterung individuell messen. Auf dieser Basis können die Entscheidungen über die Wiederholung der Therapiezyklen getroffen werden.
Labor-Basisprogramm
Vor jedem Behandlungszyklus sollte die quantitative IgG-Bestimmung in Erwägung gezogen werden (relative Kontraindikation IgG-Spiegel < 4 g/l; absolute Kontraindikation < 2 g/l). Differential-Blutbild sowie Leberparameter (GOT, GPT, Bilirubin, AP) und Nierenwerte (Kreatinin) sollten in Abhängigkeit der Begleiterkrankungen und Therapien regelmäßig erfolgen (fakultativ).
Impfungen
Spezifische Impfungen vor Therapieinitiierung sind nicht obligat erforderlich (siehe auch weiter unten).
Während der Therapie
Myasthene Krisen, die unter Efgartigimod-Therapie auftreten, können mittels IVIG, Plasmapherese (PE) oder Immunadsorption (IA) behandelt werden. Es liegen weder aussagekräftige Daten zu Kombinationstherapien von Efgartigimod mit Apherese- oder IVIG-Behandlung noch zum sinnvollen zeitlichen Abstand zwischen Apherese/IVIG-Therapie und Efgartigimod-Gabe vor. Aus mechanistischen Erwägungen heraus muss nicht nur mit einer beschleunigten Elimination von Efgartigimod gerechnet werden, sondern auch mit einer stärkeren IgG-Reduktion, die dann ggf. laborchemisch monitoriert werden muss. In der Regel sollte auf die PE/IA verzichtet und eher IVIG eingesetzt werden.
Besondere Hinweise
Schwangerschaft und Stillzeit
- Grundsätzlich sollte Efgartigimod während der Schwangerschaft und Stillzeit nicht angewendet werden. Gemäß der EMA-Empfehlungen sind Frauen im gebärfähigen Alter auf die Notwendigkeit einer wirksamen Empfängnisverhütung während der Behandlung hinzuweisen (obligat).
-
Bisher liegen keine klinischen Daten zur Anwendung von Efgartigimod bei Schwangeren vor. Eine unerwartete Schwangerschaft unter Efgartigimod ist keine zwingende Indikation für einen Schwangerschaftsabbruch. Es ist aufgrund des Wirkmechanismus von Efgartigimod mit einer Senkung der passiven Immunität des Fetus auszugehen. Ob ein Abbruch der Efgartigimod-Therapie bei Schwangerschaft erfolgen muss, bleibt eine Einzelfallentscheidung unter Nutzen-Risiko-Abwägung. - Sollte nach einer kritischen Nutzen-Risiko-Analyse im Rahmen einer individuellen Therapieentscheidung die Behandlung während einer Schwangerschaft für notwendig erachtet werden, wird zu einer strengen Überwachung von Mutter und Fetus entsprechend den lokalen Leitlinien geraten.
Impfungen
Impfungen sollten nach STIKO-Empfehlungen für Erwachsene durchgeführt werden. Spezifische Impfempfehlungen vor Therapiebeginn mit Efgartigimod liegen nicht vor. Impfungen mit Lebendimpfstoffen sind bei Patienten mit Autoimmunerkrankungen unter immunsuppressiver Therapie grundsätzlich kontraindiziert. Es liegen keine Daten über die Sicherheit von Lebendimpfstoffen unter Efgartigimod-Therapie vor. Erste Daten zeigen, dass Patienten mit gMG während der Behandlung mit Efgartigimod weiterhin in der Lage sind, schützende Titer nach Impfung zu bilden, unabhängig vom Zeitpunkt der Impfungen im Verhältnis zu den Efgartigimod-Zyklen oder der Art des Impfstoffs*.
Infektionen
Aufgrund des erhöhten Infektionsrisikos ist eine erhöhte Wachsamkeit erforderlich. Bei Symptomen von Infektionen unter Efgartigimod sind unverzüglich Maßnahmen zu Diagnostik und Therapie einzuleiten.
Dauer der Therapie
MG ist eine chronisch verlaufende Erkrankung, weshalb eine langfristige Immuntherapie grundsätzlich empfohlen wird. Aus den bisherigen Studien liegen Daten zu Therapieverläufen von mehr als 4 Jahren vor, die keine Hinweise auf Sicherheitsrisiken geben, die eine lebenslange Therapie ausschließen. Die Therapie mit Efgartigimod sollte im Verlauf regelmäßig reevaluiert werden.
Patientenaufklärung
Die DMG stellt einen Patientenaufklärungsbogen zur Therapie mit Efgartigimod für Sie bereit. Diesen können Sie hier herunterladen.
Fußnoten
* Jeffrey T. Guptill, John W. Sleasman, Sophie Steeland, Magdalena Sips, Deborah Gelinas, Hans de Haard, Antoine Azar & Kevin L. Winthrop (2022) Effect of FcRn antagonism on protective antibodies and to vaccines in IgG-mediated autoimmune diseases pem-phigus and generalised myasthenia gravis, Autoimmunity, 55:8, 620-631, DOI: 10.1080/08916934.2022.2104261.
Autoren
Diese Empfehlung wurde von der Deutschen Myasthenie Gesellschaft e. V. unter der Federführung folgender Autoren erstellt:
- Dr. med. Frauke Stascheit
Klinik für Neurologie, Charité – Universitätsmedizin Berlin
Neuroscience Clinical Research Center - Univ.-Prof. Dr. med. Andreas Meisel
Klinik für Neurologie, Charité- Universitätsmedizin Berlin
Neuroscience Clinical Research Center - Univ.-Prof. Dr. med. Jan Lünemann
Klinik für Neurologie, Universitätsklinikum Münster
Autoren
Dr. med. Frauke Stascheit
Klinik für Neurologie, Charité – Universitätsmedizin Berlin
Neuroscience Clinical Research Center
Univ.-Prof. Dr. med. Andreas Meisel
Klinik für Neurologie, Charité- Universitätsmedizin Berlin
Neuroscience Clinical Research Center
Univ.-Prof. Dr. med. Jan Lünemann
Klinik für Neurologie, Universitätsklinikum Münster